유럽 의료기기 인증
인증목적
의료기기는 유럽 연합으로의 판매를 위하여 유럽 의료기기 규정에 따라 적합함을 나타내는 CE 마크를 부착해야 합니다.
인증개요
새롭게 개정된 유럽 의료기기 규정인 Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 (이하 MDR)은 유럽 연합 내에서 판매 되는, 인간을 대상으로 하는 의료기기 및 해당 의료기기의 액세서리를 규제하는 규격입니다. MDR은 기존에 적용되던 의료기기 지침인 Council Directive 93/42/EEC on Medical Devices (이하MDD) 및 전기 전자 이식형 의료기기 지침인 Council Directive 90/385/EEC on Active Implantable Medical Devices (AIMDD)를 대체하게 됩니다. MDR은 2020년 5월 26일부터 완전히 시행 될 계획이었으나, 전 세계적인 코로나 바이러스의 유행으로 인하여 적용날짜가 1년간 연기되어 2021년 5월 26일부터 완전히 적용 되었습니다. 더하여, 체외 진단 의료기기(Invitrodiagnostic medical devices)의 새로운 규정인 Regulation (EU) 2017/746 (IVDR)은 다가오는 2022년 5월부터 적용되며, 기존의 체외 진단 의료기기지침인 Directive 98/79/EC (IVDD)를 대체하게 됩니다.
MDD와 MDR 차이
- Basic UDI-DI 추가
- UDI 시스템 (UDI System) 도입
- 유럽의료기기데이터베이스 (EUDAMED) 도입
- 정기안전업데이트보고서 (Periodic Safety Update Report, PSUR) 추가
- 안전성및임상적성능요약 (Summary of Safety and Clinical Performance, SSCP) 추가
- 임플란트카드 (Implant Card) 추가
- 인증기관(Notified body, NB)에 대한 엄격한 요구사항 적용
- 의료기기등급분류의확장
의료기기 등급 분류
의료기기의 등급은 기기의 의도된 목적과 내재된 위험을 고려하여 MDR의 부속서 VIII 등급 분류 규칙에 따라 진행됩니다.
등급이 높은 의료기기 일수록 내재된 위험이 높음을 의미합니다.
- I등급 (멸균, 측정, 재사용 가능한 수술용 기기 포함)
- IIa등급
- IIb등급
- III등급
적합성 평가 절차
의료기기의 등급별 해당하는 적합성 평가 절차에 따라 요구사항의 준수가 입증 된 경우, 의료기기 제조자는 제 19조에 따라 EU 적합성선언(EU Declaration of Conformity, EU DoC)을 작성하고 제 20조에 따라 CE 마크를 부착해야 합니다. (주문제작 또는 임상조사용 기기는 별도)
하기 적합성 평가 절차는 의료기기의 특성에 따라 다르게 적용될 수 있습니다.
-
- I 등급 :Annex II → Annex III → Annex IV → Annex V
-
- IIa 등급 :a) Chapters I and III of Annex IX → Section 4 of Annex IX → Annex IV → Annex V
b) Part A of Annex XI → Annex II → Annex III → Annex IV → Annex V
c) Part B of Annex XI → Annex II → Annex III → Annex IV → Annex V -
- IIb 등급 :a) Chapters I and III of Annex IX → Section 4 of Annex IX
→Section 5 of Annex IX → Section 6 of Annex X →Annex IV → Annex V
b) Annex X → Part A of Annex XI → Section 5 of Annex IX → Section 6 of Annex X → Annex IV → Annex V
c) Annex X → Part B of Annex XI → Section 5 of Annex IX → Section 6 of Annex X → Annex IV → Annex V -
- III 등급 :a) Chapters I and III of Annex IX → Section 4 of Annex IX → Annex IV → Annex V
b) Annex X → Part A of Annex XI → Annex IV → Annex V
c) Annex X → Part B of Annex XI → Annex IV → Annex V
의료기기기술문서항목
| 번호 | 국문 | 영문 |
|---|---|---|
| 1 | 의료기기에 대한 설명 및 기기의 사양 (기기의 의도된 목적, 적용 대상 질환, 사용 대상 환자, 사용시 주의사항 등) |
Device description and specification (intended purpose, medical conditions, intended patient population, warnings, etc.) |
| 2 | 이전 세대 및 유사한 세대의 의료기기에 대한 참조 | Reference to previous and similar generation of the medical device |
| 3 | 제조자가 제공하는 정보 (라벨 및 사용설명서) | Information to be supplied by the manufacturer (Label and the instructions for use) |
| 4 | 의료기기의 설계 정보 및 제조 정보 | Design and manufacturing information |
| 5 | 일반적인 안전성 및 성능 요구사항 | General safety and performance requirements |
| 6 | 이득 - 위험분석을 포함한 위험관리 | Benefit-risk analysis and risk management |
| 7 | 의료기기의 사전 임상 안전성 | Pre-clinical safety of the medical device |
| 8 | 생체 적합성 테스트 | Biocompatibility of the medical device |
| 9 | 물리적, 화학적, 미생물학적 테스트 | Physical, chemical and microbiological tests |
| 10 | 전기안전성 및 전자기적 적합성 테스트 | Electrical safety and electromagnetic compatibility tests |
| 11 | 소프트웨어 검증 및 유효성 확인 | Software verification and validation |
| 12 | 의료기기의 유효기간을 포함한 안정성 시험 | Stability test including shelf life |
| 13 | 임상평가계획서 및 보고서와 업데이트 | Clinical evaluation plan, report, and its updates |
| 14 | PMCF 계획 및 평가보고서 | PMCF(Post-market clinical follow-up) plan and evaluation report |
| 15 | 안전성 및 임상성능(SSCP)의 요약 | Summary of Safety and Clinical Performance(SSCP) |
| 16 | 품질관리 문서화 | Quality management documentation |
| 17 | 사후 감시계획 및 보고서 | Post-market surveillance plan and report |
| 18 | 의료기기 Vigilance | Vigilance of the medical device |
| 19 | EU 적합성 선언서(DoC) | EU Declaration of Conformity |
의료기기의특성에따라요구되는사항이달라질수있습니다.
예)전기전자의료기기여부,의료기기와환자와의접촉여부, 이식용의료기기여부등
예)전기전자의료기기여부,의료기기와환자와의접촉여부, 이식용의료기기여부등
필요자료
- 영문 사업자등록증
- 제품 사양서 또는 사용자설명서 등 제품 설명에 대한 문서
- 라벨 등의 표시사항
- 기기설계 및 공정관련문서 (설계도, 공정도등)
- 관련 시험성적서
- 그외 요청되는 문서 등
진행절차
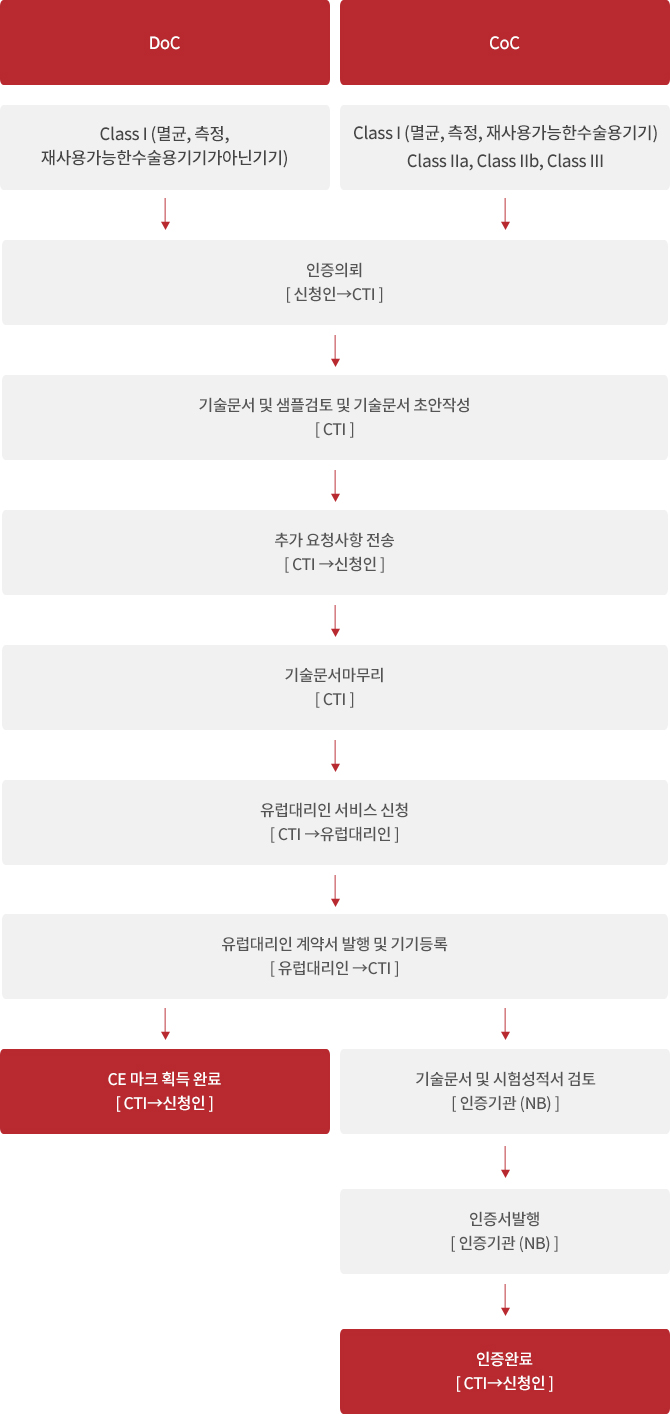
* 부적합시 부적합 사항 (문서 또는 샘플)의 보완진행 필요.
비고사항
- 유효기간
- 인증기관 (NB)에 의하여 발행된 CE 인증서의 경우, 일반적으로 3년의 유효기간을 가집니다. 그러나 의료기기의 특성이나 위험정도에 따라 변동이 있을 수 있습니다.
하단영역
(주)씨티아이인증원
대표 고현미
사업자등록번호 : 729-88-00808
경기도 광명시 일직로43, A동 1712호(일직동, 지아이디씨) 우편번호:14353
TEL : 02-6914-5600
FAX : 02-6914-5603
E-mail : hskang@cticert.co.kr
경기도 광명시 일직로43, A동 1712호(일직동, 지아이디씨) 우편번호:14353
TEL : 02-6914-5600
FAX : 02-6914-5603
E-mail : hskang@cticert.co.kr
찾아오시는 길
(주)씨티아이인증원의 오시는 길을 안내합니다.
GO
Copyright (C) 2021. CTI. All Rights Reserved.