FDA

인증목적
의료기기는 미국으로의 판매를 위하여 FDA에 시설 등록 및 기기 등재가 되어야 합니다.
미국 내에서 사용되기 위한 의료기기의 생산 및 유통과 관련된 시설의 소유자/운영자는 해당 시설을 매년 FDA에 등록해야 합니다.
이러한 프로세스를 시설 등록 (Establishment registration)이라고 합니다.
이에 더하여, 일반적으로 등록 대상인 시설의 의료기기는 FDA에 등재가 요구됩니다. 이러한 프로세스를 기기 등 재 (Device listing)라고 합니다.
미국 내에서 사용되기 위한 의료기기의 생산 및 유통과 관련된 시설의 소유자/운영자는 해당 시설을 매년 FDA에 등록해야 합니다.
이러한 프로세스를 시설 등록 (Establishment registration)이라고 합니다.
이에 더하여, 일반적으로 등록 대상인 시설의 의료기기는 FDA에 등재가 요구됩니다. 이러한 프로세스를 기기 등 재 (Device listing)라고 합니다.
인증개요
FDA는 기기의 위험에 따라 의료기기를 세 개의 등급 (Class I, Class II, Class III)으로 분류합니다.
등급이 높을수록 위험이 높고, 규제가 강화되며, 기기의 등급에 따라 다른 제출 유형 (Submission Type)이 적용됩니다.
- Class I : 대부분의 I 등급 기기는 510(k)가 면제됩니다. (예: 안경, 칫솔등)
절차가 모두 완료되면 해당 의료기기는 “FDA 등재(listed)” 된 기기입니다. - Class II : 대부분의 II 등급 기기는 510(k)의 대상입니다. (예: 카테터, 산소측정기등)
절차가 모두 완료되면 해당 의료기기는 “FDA 확인(cleared)” 된 기기입니다. - Class III : 대부분의 III 등급 기기는 시판전승인 (Premarket Approval, PMA)의 대상입니다. (예: 심장박동조율기, 인공관절등)
절차가 모두 완료되면 해당 의료 기기는 “FDA 승인(approved)” 된 기기입니다.
이에 더하여, FDA는 각 의료기기를 제품코드 (Product code)로 분류합니다. 의료기기 제품코드 데이터베이스에서 제품코드를 검색하여 각 의료기기에
해당하는 제출 유형 (510(k) 면제인지, 대상인지, PMA 대상인지 등)을 확인할 수 있습니다.
Class 1 – 510(k) 면제
I 등급 의료기기는 FDA의 의료기기 등급 분류 중 위험도가 가장 낮은 등급입니다.
대부분의 I 등급 의료기기는 510(k) 면제이므로, 시설 등록 및 기기 등 재 완료 후 의료기기의 미국 내 판매가 가능합니다.
그러나, 모든 I 등급 기기가 510(k) 면제에 해당하는 것은 아니므로, 판매를 원하는 의료기기의 제품코드 검색을 통하여 제출 유형의 정확한 확인이 필요합니다.
대부분의 I 등급 의료기기는 510(k) 면제이므로, 시설 등록 및 기기 등 재 완료 후 의료기기의 미국 내 판매가 가능합니다.
그러나, 모든 I 등급 기기가 510(k) 면제에 해당하는 것은 아니므로, 판매를 원하는 의료기기의 제품코드 검색을 통하여 제출 유형의 정확한 확인이 필요합니다.
Class 2 – 510(k) 대상
510(k)는 해당 의료기기와 기존 의료기기 (predicate device)의 본질적 동등성 (substantially equivalent)을 입증하기 위해 FDA에 제출하는
시판 전 제출 (premarket submission)입니다. 본질적 동등성은 새로운 기기가 기존의 기기만큼 안전하고 효과적이라는 것을 의미합니다.
510(k) 제출본의 내용은 다음을 포함합니다.
- 의도된 사용(intended use) 및 사용시 적응 증(indications for use)
- 510(k) 요약
- 기기설명
- 라벨링
- 성능시험 자료등
510(k) 제출본이 FDA에 제출된 이후, FDA는 허용 검토 (acceptance review), 실질 검토 (substantive review), 상호 검토 (interactive review)의 단계를 거치며
본질적 동등성을 검토합니다.
소기업 감면 프로그램 (Small Business Determination Program)
고객사가 소기업에 해당할 경우, 510(k), PMA, BLA, De Novo 등 비용이 발생하는 절차 진행 시 비용 감면을 받을 수 있습니다.
단, 시설 등록 시 발생하는 비용은 소기업 감면의 대상이 아닙니다.
소기업의 정의 : 계열사를 포함하여, 가장 최근과 세 연도의 총 수입 또는 총 매출이 1억달러 (한화약1,200억) 이하인 기업.
필요자료
-
시설등록 및 기기등재
(Establishment registration and
device listing)- - 영문사업자등록증
- - 최근 3개년재무제표
- - 법인등기부등본
- - 주주명부
- - 최근 3개월 4대보험사업장고지내역서
- - 대표자, 제조업체정보
- - 제품정보 (제품명, 제품코드등)
-
510(k) 시판전신고
(Premarket notification, 510(k))- - 시설 등록 및 기기등재에 필요한 서류
(상기내용확인) - - 제품 사양서/제품표준서/기술문서/사용자 설명서 등 제품설명에대한 문서
- - 라벨 등의 표시 사항
- - 관련 시험성적서
- - 그외 510(k) 제출본의 작성을 위하여 요청되는 문서등
- - 시설 등록 및 기기등재에 필요한 서류
-
소기업신청
(Small business determination
(SBD) program)- - 세금관련 증빙문서
- - Form FDA 3602A (국세청 방문 필요)
진행절차
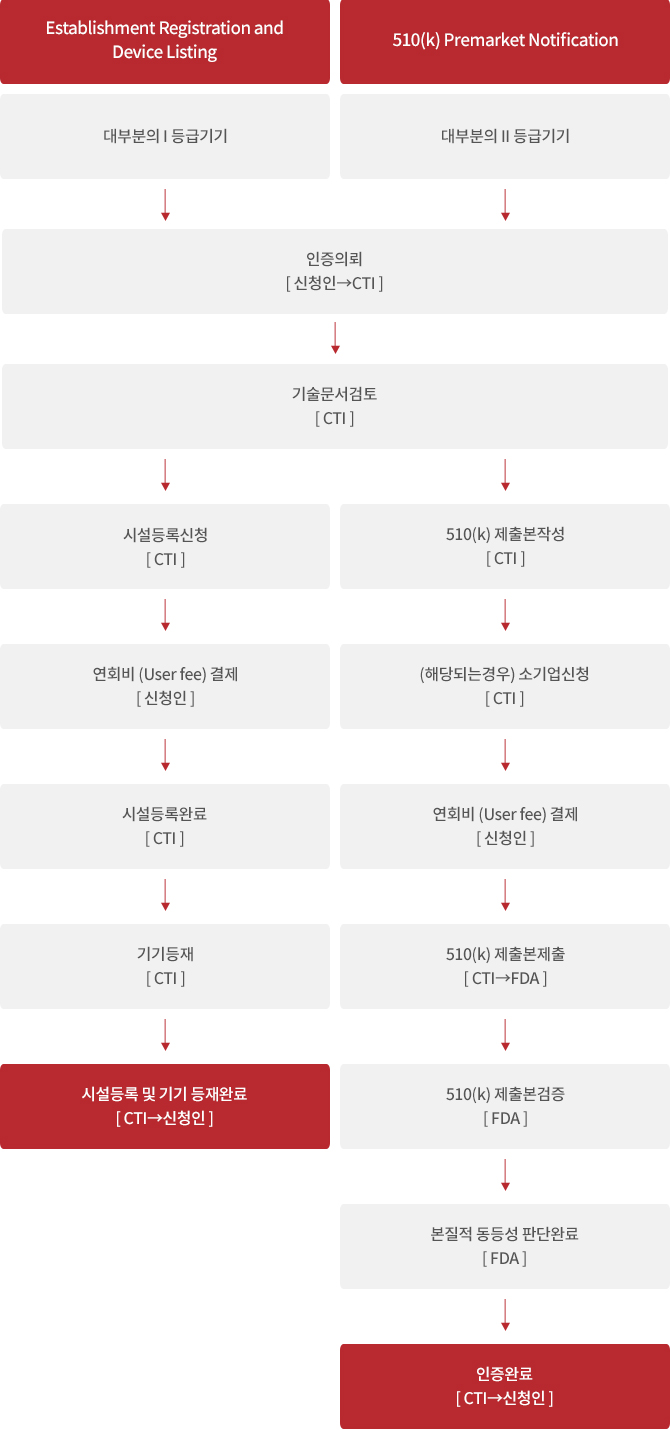
* 부적합시 부적합 사항 (문서 또는 샘플)의 보완진행 필요.
이에 따라 소요기간이 연장될 수 있습니다.
이에 따라 소요기간이 연장될 수 있습니다.
비고사항
- 회계년도
- FDA에서 비용 지불의 기준인 FY는 회계년도 (FY, Fiscal Year)를 의미합니다. 매회계연도는 전년도 10월1일부터 당년도 9월30일까지 입니다.
* 예시) FY 2022의 시설등록비용을 납부하시는 경우
FY 2022 유효기간: 2021년 10월 1일 ~ 2022년 9월 30일
FY 2022 연간비용: $5,672
FY 2022 유효기간: 2021년 10월 1일 ~ 2022년 9월 30일
FY 2022 연간비용: $5,672
하단영역
(주)씨티아이인증원
대표 고현미
사업자등록번호 : 729-88-00808
경기도 광명시 일직로43, A동 1712호(일직동, 지아이디씨) 우편번호:14353
TEL : 02-6914-5600
FAX : 02-6914-5603
E-mail : hskang@cticert.co.kr
경기도 광명시 일직로43, A동 1712호(일직동, 지아이디씨) 우편번호:14353
TEL : 02-6914-5600
FAX : 02-6914-5603
E-mail : hskang@cticert.co.kr
찾아오시는 길
(주)씨티아이인증원의 오시는 길을 안내합니다.
GO
Copyright (C) 2021. CTI. All Rights Reserved.