Korea MFDS Certificate
Domestic Medical Device (MFDS)

Purpose
The purpose of 『Domestic Medical Device Notification, Certification or Approval』 is to promote efficient management of medical devices and to contribute to the improvement of public health by stipulating matters concerning manufacturing, import, repair, and sales.
(No. 13698 “Medical Device Act”, No. 2015-114 “Regulation on Medical Device Approval Report Review, Etc.”)
(No. 13698 “Medical Device Act”, No. 2015-114 “Regulation on Medical Device Approval Report Review, Etc.”)
Overview
In accordance with『Domestic Medical Device Notification, Certification, or Approval』, a person who intends to manufacture, import, repair, sell, or lease medical devices shall report, certify, and obtain Approval for the relevant medical device to sell medical devices in Korea.
Target
- 「Medical device handler」
- A “medical device handler” refers to any of the following parties that handle medical devices as part of its business and that have obtained a license or filed a report under the Medical Device Act;
- A manufacturer of medical devices
- An importer of medical devices
- A repairer of medical devices
- A seller of medical devices
- A lessor of medical devices
Required Documentation
-
Notification of manufacturing (import)
Class I medical device- • Notification (Attachment No. 7 Form of the Enforcement Rule of the Medical Devices Act)
- • Purpose of use, etc. (Attached table 1 of Regulations for Product Classification of Medical Device and Class by Product)
- • In case of a consignment of the entire process: Documents proving the conditions of the consignee (documents that can prove whether he/she is a person who can be entrusted with the entire manufacturing process)
- • For item category notification: When notifying manufacturing (import), mark in the “Items” column of the notification
- ※For medical devices that are not essentially equivalent in structure, principle, performance, the purpose of use, and method of use to those that have already been approved or notified among Class I, you shall apply for Approval to the Ministry of Food and Drug Safety.
-
Notification of manufacturing (import) modification
Class I medical device- • Notification (Attachment No. 7 Form of the Enforcement Rule of the Medical Devices Act)
- • In case of a consignment of the entire process: Documents proving the conditions of the consignee (documents that can prove whether he/she is a person who can be entrusted with the entire manufacturing process)
- • In case of general modification: If there is a change in the product notified for manufacturing (import), attach the 'Minor Kor_Changes Report' as minor Kor_Changes and submit it to the head of the competent regional Food and Drug Administration within 30 days or from the last day of the month preceding the date of initial Approval or notification to the last day of the month every year
- ※ If the registration is changed on the medical device e-complaint site, the modification is considered to have been notified
- • In case of modification due to transfer or acquisition: Treated as an occasional report of minor change
- * When processing a report of minor changes due to transfer or acquisition, the name of the manufacturer and product name change is processed together and submission of the transfer or acquisition agreement (original) is required.
-
Equivalent product
Class II medical device- • Application for manufacturing (import) certification • Approval of medical devices
(Attachment No. 3/5 form of the Enforcement Rule of the Medical Devices Act) - • Materials equivalent to technical documents of licensed products
- • A copy of the compliance certificate to Medical Device Manufacturing and Quality Control Standards (GMP) for the item or the identical item
- ※ In accordance with Article 6 (4) of the “Medical Device Act”, a person who attempts to obtain a manufacturing license or manufacturing certification or notify manufacturing after January 29, 2016 shall prepare the necessary facilities and manufacturing and quality control system in advance to apply for or notify a license or certification.
- • Manufacturing • sales certificate, User’s manual
- • Manufacturer's certificate (notarization)
- • Application for manufacturing (import) certification • Approval of medical devices
-
Recognized substantial equivalent products
Class II medical device- • Application for manufacturing (import) certification • Approval of medical devices
(Attachment No. 3/5 form of theEnforcement Rule of the Medical Devices Act) - • A test and inspection report issued by a testing and inspection institution designated by the Minister of Food and Drug Safety
- ※A test report proving that it is suitable for the product announced as equivalent
- ※A test report issued by a testing and inspection institution designated by the Minister of Food and Drug Safety
- • A copy of the compliance certificate to Medical Device Manufacturing and Quality Control Standards (GMP) in the item or the identical item group
- ※ In accordance with Article 6 (4) of the “Medical Device Act”, a person who attempts to obtain a manufacturing license or manufacturing certification or report manufacturing after January 29, 2016 shall prepare the necessary facilities and manufacturing and quality control system in advance to apply for or report a license or certification.
- • Documents proving the conditions of consignee (limited to entire process consignment)
- • Comparison table of essentially equivalent items
- • Application for manufacturing (import) certification • Approval of medical devices
-
Class III and IV medical devices
- • Application for Approval of medical device manufacture(import)
(Attachment No. 3 form of the Enforcement Rule of the Medical Devices Act) - • Notification of examination results such as technical documents, technical documents, and data on clinical trials
- ※ The notification of examination results such as technical documents, etc. shall be within two years from the date of issuance
- • Documents proving the conditions of consignee (limited to entire process consignment)
- • Application for Approval of medical device manufacture(import)
Process
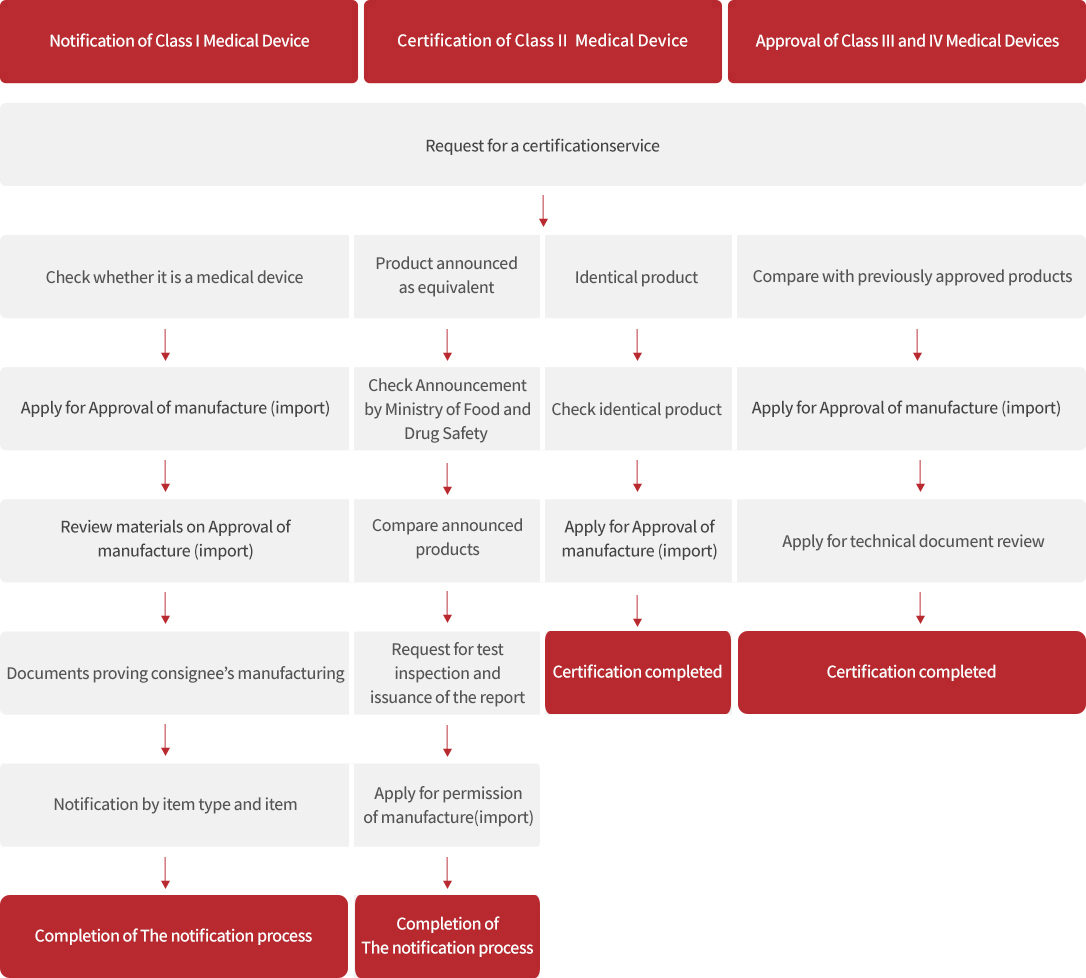
※ In case of nonconformity, the nonconformity (document or sample) needs to be supplemented.
Remarks
- Article 51(Penalty)
- A person who falls under any of the following shall be punished by imprisonment for not more than five years or by a fine not exceeding 50 million won :
- • A person who has obtained Approval or certification or made a report underParagraphs 1•2 of Article 6 or Paragraphs 1•2 of Article 15 by fraud or
other illegal means; - • A person who violates Paragraph1 of Article 26;
- • A person who has received renewal under Paragraph 3 of Article 49 by fraud or other illegal means;
Bottom area
CTI Co., Ltd
CEO Hyun Mi Ko
Business registration number : 729-88-00808
GIDC A-1712, 43, Iljik-ro, Gwangmyeong-si, Gyeonggi-do, Republic of Korea Zipcode:14353
TEL : +82 2-6914-5600
FAX : +82 2-6914-5603
E-mail : hskang@cticert.co.kr
GIDC A-1712, 43, Iljik-ro, Gwangmyeong-si, Gyeonggi-do, Republic of Korea Zipcode:14353
TEL : +82 2-6914-5600
FAX : +82 2-6914-5603
E-mail : hskang@cticert.co.kr
Visit us
CTI location
GO
Copyright (C) 2021. CTI. All Rights Reserved.